
Epi25 – redefining epilepsy genetics through exomes of 17,000 individuals
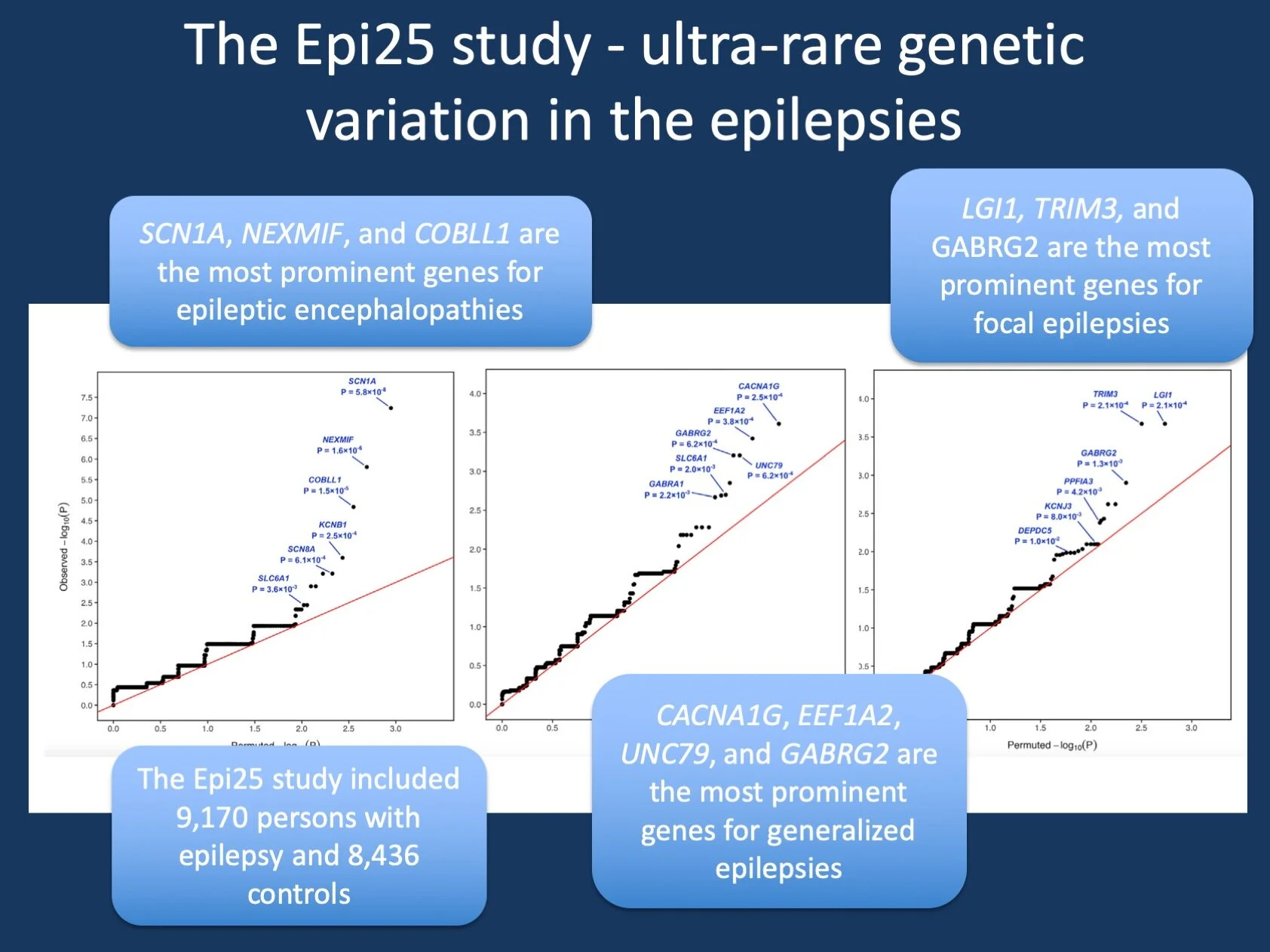
Figure 1. Burden analysis for the three phenotypic subgroups in the Epi25 study, including developmental and epileptic encephalopathies (DEE), generalized genetic epilepsies (GGE), and non-acquired focal epilepsy (NAFE). In brief, a burden analysis assesses the frequency (“burden”) of rare variants in cases and controls and assesses whether this difference is significant. The current graph (qq plot) indicates genes that deviate from the expected p-value (red line), hinting at a role in disease causation. While no single gene reached exome-wide significance, several genes are suggestive, most prominently SCN1A in DEE. Several other genes, including genes that were previously not implicated in the epilepsies, were found to carry more ultrarare variants than expected, such as COBLL1 for DEE, UNC79 for GGE, and TRIM3 for NAFE [modified from Figure 3 of the corrected proof (link) with permissions by the publisher]
Posted on August 6, 2019 by Ingo Helbig
The Epi25 study. On August 1, the Epi25 study was published in the American Journal of Human Genetics. Epi25 is the major, international effort to understand the genetics of common and rare epilepsies through exome sequencing, and our current study now presents the first results on what we can see if we look at the genetics of the epilepsies in thousands of individuals, including more than 9,000 persons with epilepsy and 8,000 controls. The Epi25 study finds that individuals with epilepsy carry more ultra-rare, deleterious variants than controls, especially in known or presumed candidate genes. This is a significant finding that tells us about the inner genetic architecture of the epilepsies beyond the role of monogenic causes. However, as with many previous studies at this scale, the first publication merely scratches the surface and provides an enormous amount of data for further studies. Here is a brief summary of the Epi25 study and some of the most prominent genes in the epilepsies that were completely unknown previously.
Epi25 analysis. The Epi25 study is a large case-control study, which is important to keep in mind when trying to understand whether known or newly identified genes are “causative”, a label that we usually reserve for monogenic disorders. The variants in Epi25 sometimes come quite close to this label as the study looked at types of ultra-rare genetic variants, which are completely absent in large control population. Especially for so-called protein truncating variants (PTVs), such as stop mutations in SCN1A or splice-site variants in STXBP1, the analysis basically looks at variants that would be flagged as pathogenic in a diagnostic context. However, as in so many cases of large-scale studies, there is no clear black and white difference in Epi25. For example, PTVs in genes for 50 dominant epileptic encephalopathies such as SCN1A are present both in cases and controls, but three times more likely (odds ratio of 3.0) in individuals with epilepsy. However, in the more than 8,000 control individuals, 21 protein truncating variants in these 50 genes for dominant epileptic encephalopathies were identified.
The Epi25 browser. This blog post won’t be able to go into the details for all genes assessed in the Epi25 study. However, the Broad Institute, the main center where sequencing and data analysis was performed for Epi25, has provided gene-specific information on a dedicated Epi25 browser (link) that allows us to take a deep dive into each specific gene in the three cohorts analyzed in Epi25, namely developmental and epileptic encephalopathies (DEE), generalized genetic epilepsy (GEE), and non-acquired focal epilepsy (NAFE). For example, for STXBP1, this browser shows that 3 individuals with epilepsy and one control individual were found to have protein-truncating variants in STXBP1. Again, as with the gNOMAD browser, be aware that the gene search field has a tricky auto-correct function that sometimes returns a different gene than you had initially asked for.
The Epi25 results. Results of the magnitude of Epi25 can only be delineated in very broad strokes in a blog post. Basically, the study finds that ultra-rare variants are more common in the various types of epilepsies than in controls. In addition, the degree to which these variants are enriched in the epilepsies increases with the predicted pathogenicity of these variants. No single gene is significant when testing for exome-wide significance, but there is a strong signal of specific gene groups, such as in genes for GABA-A receptor subunits, known epilepsy and neurodevelopmental genes, and genes that are intolerant to variation in control population that can be interpreted as “hidden disease genes”. These results are highly important as they highlight that the genetic architecture of both common and rare epilepsies has a strong contribution of many ultra-rare variants. For the overall genetic architecture of the epilepsies, this generated a multifaceted picture with contributions of common genetic variants, rare genetic variants, and monogenic causes. Understanding this picture is critically important when thinking about how we can translate genetic findings into new therapeutic strategies and finding “themes” in the dataset such as the contribution of GABA-A receptor genes is an important first step in this direction.
Genes in Epi25. Even though no single gene stood out, there are a few genes in Epi25 that are worth noting. First, SCN1A is the most prominent gene in DEE, a finding that is both expected and reassuring. However, the same strength of association for SCN1A is not found in GGE or NAFE, reinforcing that SCN1A-related epilepsies only contribute little to the genetic architecture of common epilepsies. In contrast, several genes emerge from the GGE and NAFE analysis that are surprising. The most prominent gene in GGE is CACNA1G, a gene for a voltage-activated T-type calcium channel highly expressed in the cerebellum. CACNA1Gis the causative gene for spinocerebellar ataxia-42 (SCA42) and while ion channel genes are frequently found as genetic contributors to the epilepsies, the prominence of this specific gene is unusual. For NAFE, the established LGI1 gene shares its role as the most prominent gene with TRIM3, a gene that was not previously implicated in disease. TRIM3 encodes the Tripartite Motif Containing 3 protein, a brain-expressed protein of unknown function. Finally, the COBLL1 gene emerges for the DEE analysis as the third most prominent gene. COBLL1 encodes a protein with the unusual name of Cordon-Bleu WH2 Repeat Protein Like 1, also a brain-expressed gene of unknown function. Of note, COBLL1 is the genetic neighbor of SCN1A, SCN2A, and SCN3A on chromosome 2q24.3.
What you need to know. The Epi25 study is the first step into a new era of epilepsy genetics, moving away for the de novo paradigm in epileptic encephalopathies to a broader assessment of risk variants in both common and rare epilepsies. Epi25 highlights the important role of rare variants that form a complicated puzzle contributing to the genetic architecture of the epilepsies and emphasizes the need to look at gene groups such as genes for inhibitory GABA-A receptors as no single gene is a major contribution. Finally, the current Epi25 study is just the beginning. As implied by the name of the collaborative (“twenty-five” for 25,000), further sequencing and analysis is ongoing and our current publication only reports the results of the first two years of recruitment and sequencing. In addition, Epi25 has already become the hub for several spin-off projects, including studies of other types of genetic variation, candidate genes, or (as the case of our group), new strategies to analyze the complex phenotypes through computational methods.